
近年来,血管外科领域取得了引人注目的进展,无论是技术革新、设备升级还是治疗方法优化,都展现出了蓬勃发展的态势。本期内容是由来自复旦大学附属中山医院的符伟国教授带来《医生改良腔内移植物(PMEG)国内外现状》的精彩内容,欢迎阅读与分享!

背景
心血管疾病已跃居为我国居民首要致死病因,其中主动脉疾病作为其核心构成部分,其影响尤为显著。据国内权威数据揭示,主动脉夹层的死亡率居高不下,维持在6%至8%之间,而2019年,主动脉瘤导致的死亡人数更是高达1.7万人。
随着医疗科技的飞速跃进,主动脉疾病的治疗策略正经历着深刻的变革,从巨创的开放手术,逐步过渡为微创的腔内手术。近年来,我国主动脉疾病腔内治疗的应用范围与频率均呈现出显著增长态势。从2017年的21,320例治疗案例,到2021年激增至46,651例,增幅惊人,达到了118.1%,彰显了腔内治疗技术在我国的广泛接受度与快速发展。
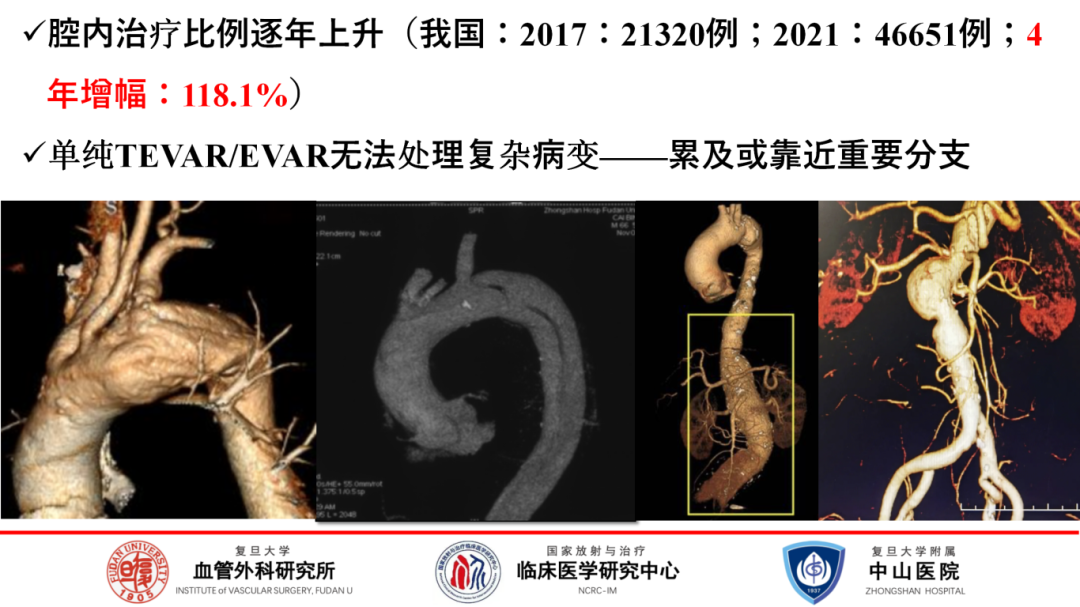
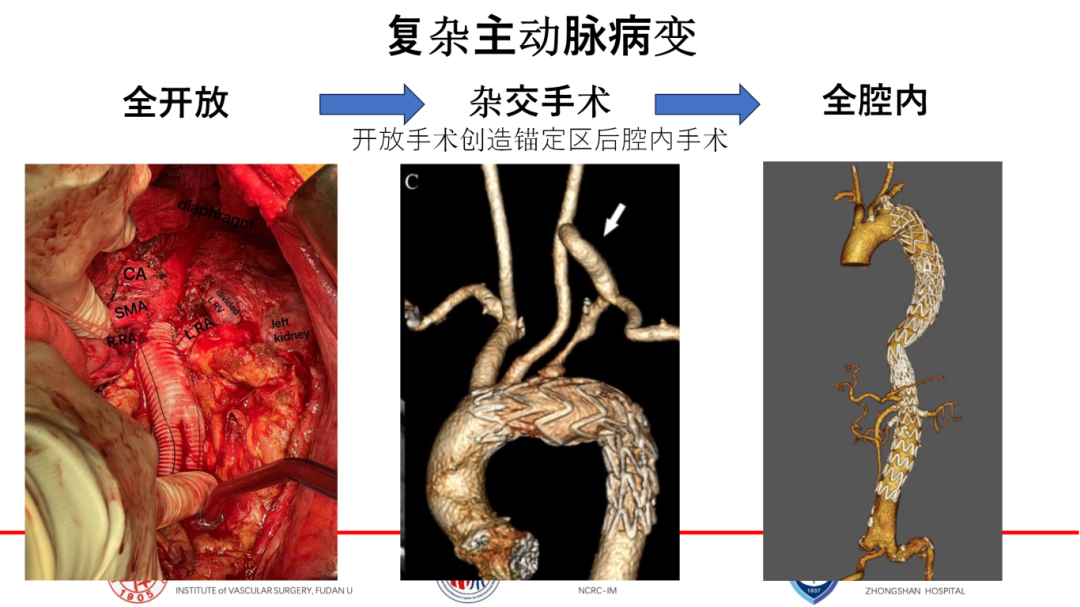
然而面对部分复杂病例,如病变累及重要分支的情况,单纯依赖TEVAR/EVAR技术仍显力不从心。为此不断创新,从最初的开放手术重建分支,到采用杂交手术策略,即先创造足够的锚定区域,再行支架覆盖,直至如今探索全腔内治疗手段,每一步都凝聚着医学智慧与不懈追求。
全腔内技术探索之旅
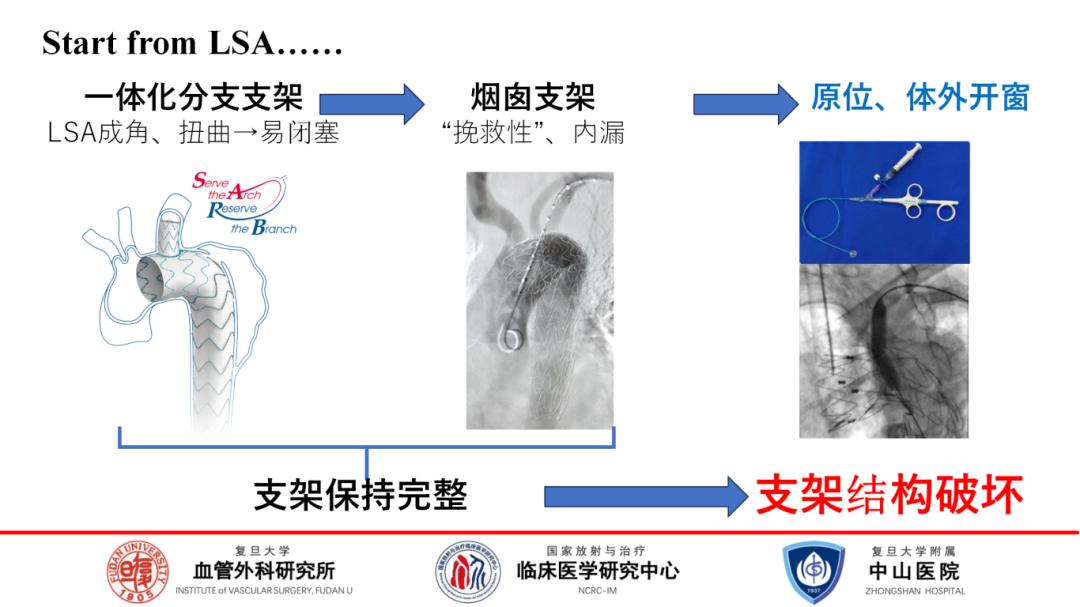
全腔内治疗技术的探索之旅,始于对左锁骨下动脉(LSA)重建的深入探索。初期,寄望于一体化分支支架来保全LSA,然而面对LSA成角扭曲挑战,该方案常导致分支闭塞。随后,烟囱支架等平行支架技术应运而生,力求重建LSA,但这些“挽救性支架”间易产生GUTTER,进而增加内漏风险。直至今日,迎来了原位开窗与体外开窗技术,它们标志着对支架结构创新性破坏的同时,也引发了关于支架完整性与功能平衡的讨论。
追溯改制支架的历史,1999年Browne首次在动物实验中报道其成功保留肾动脉血流的壮举,为后来发展奠定了基础。2001年,Anderson的开创性工作展示了开窗支架在治疗肾周腹主动脉瘤(AAA)患者中的潜力。2008年,日本学者Yokoi等人通过Najuta定制开窗支架的临床试验,进一步推动了体外预开窗技术的发展。


2012年,是主动脉腔内治疗的重要里程碑。Starnes不仅提出了PMEG(预成型模块化内移植物)概念,还率先获得了FDA批准,开展肾周AAA治疗的前瞻性研究,其成果彰显了PMEG的临床价值。同年,FDA批准了首个厂家定制成品分支支架Zenith Fenestration(Cook),为临床提供了又一有力工具。


国内外PMEG现状
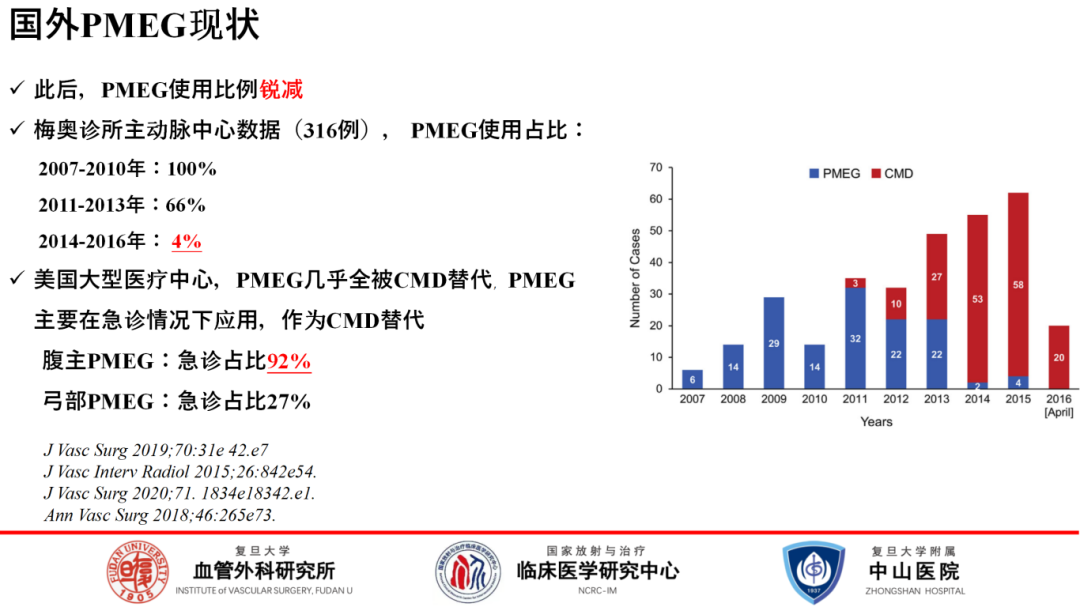
然而,随后在美国,PMEG的使用率逐渐下降,被CMD(定制设备)广泛取代,仅在紧急情况下,如腹主动脉瘤破裂时,PMEG仍保留一席之地。
随着医学指南的更新,PMEG的适应症也在演变。2019年ESVS指南虽未将PMEG列为肾周AAA的一线治疗,但在紧急情况下仍被推荐。2024年最新版ESVS指南,PMEG的适应症有所放宽,同时考虑了患者的个人偏好。
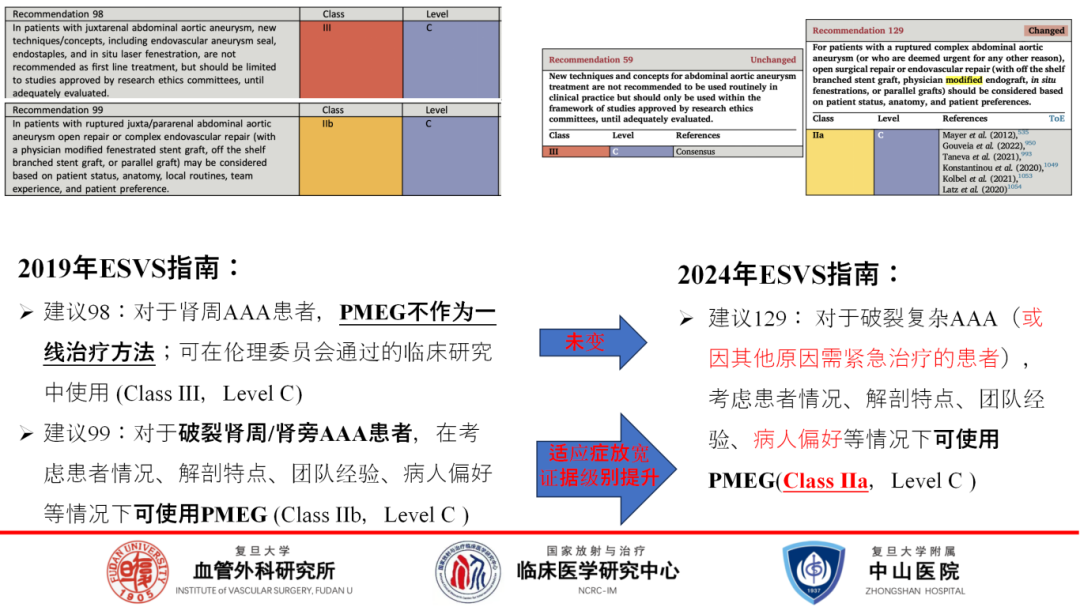
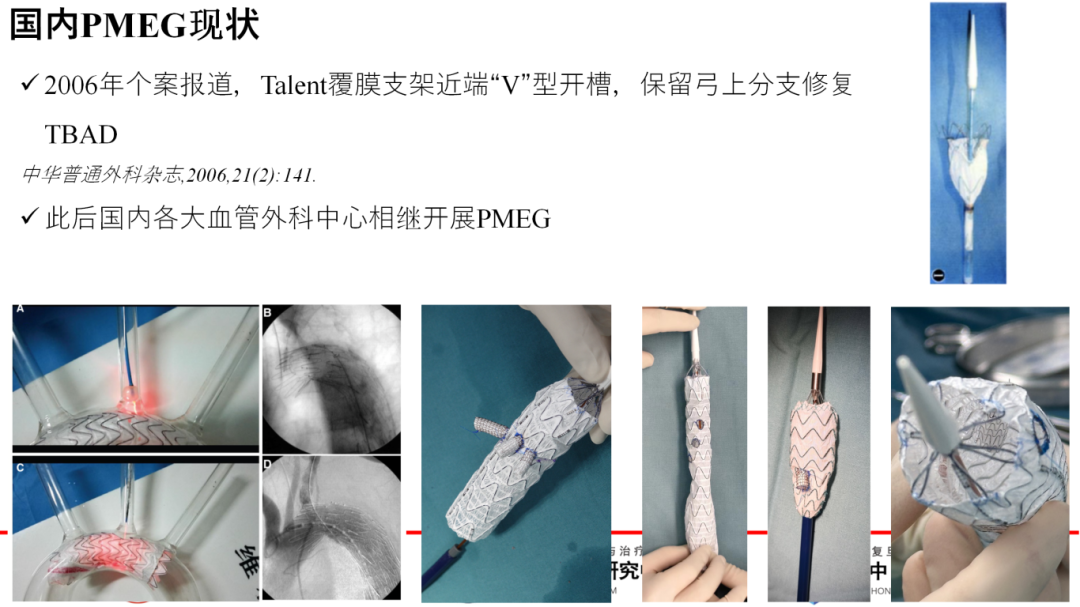
在国内,PMEG的引入与发展同样令人瞩目。2006年首例PMEG报道,通过在Talent支架上进行V形开槽,保留弓上分支血供,标志着我国在复杂主动脉病变治疗上的新突破。此后,PMEG技术在国内迅速普及,各大血管外科中心纷纷采用,包括体外开窗、原位开窗、内外分支等多种策略,共同推动我国主动脉腔内治疗技术的全面发展。
至今,尽管精确统计改制支架的具体数量存在难度,但依据HQMS数据,以TEVAR为例,按41%的病例需锚定于2区,排除一体式支架后,估算改制支架的占比约为15%至20%。
与PMEG形成鲜明对比的是CMD,遗憾的是,我国尚未有CMD产品上市,故PMEG目前是国内治疗复杂主动脉病变的主导方案。尽管研究表明,PMEG与CMD在内漏、再干预等关键指标上无显著差异,CMD却以其高度个性化匹配患者解剖结构及生产企业的严格质量控制为显著优势。相比之下,PMEG虽无需漫长等待,却面临技术门槛较高及质控标准不统一、认可度不足的挑战,且其超适应症使用的伦理问题亦备受争议。值得注意的是,尽管CMD目前以手工改制为主,由资深技术人员精细打造,且改制后经历严格消毒与二次质检,这一流程确保了产品的安全性与有效性,是PMEG所难以比拟的。

综上所述,在国内现状下,PMEG虽被视为复杂主动脉病变的首选疗法,但公众主要关切聚焦于两点:一是改制过程是否会影响支架原有性能;二是现行国内外法规如何界定PMEG的合规性,确保患者权益得到充分保障。
PMEG对于支架性质的影响
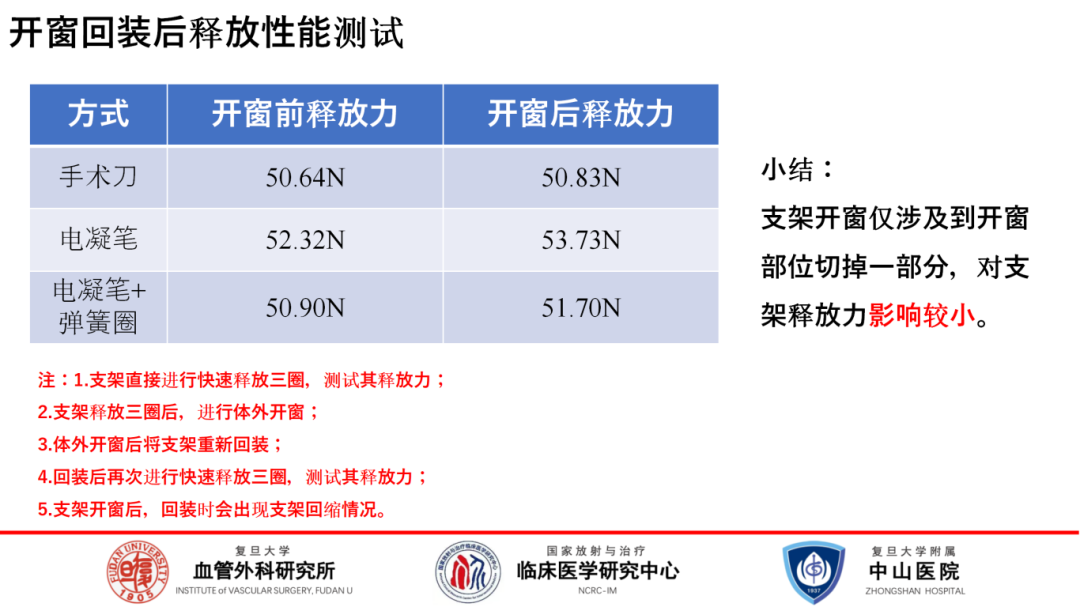
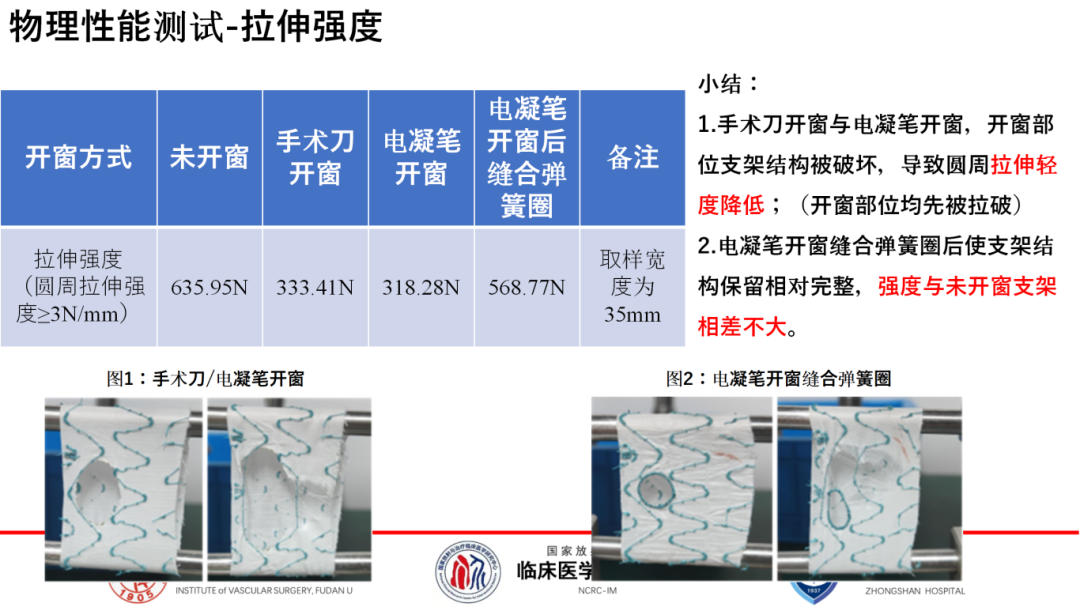
首先聚焦于改制后支架性质的潜在变化。本研究系统地对比了未开窗支架与通过手术刀开窗、电凝笔开窗及电凝后锁边开窗处理的支架。在支架回装后的释放性能测试中,各处理组之间展现出相似的性能,表明开窗操作对支架的释放力影响微乎其微。然而,在拉伸强度评估上观察到了一个显著的区别,手术刀和电凝笔开窗后的支架,其拉伸强度均出现了明显下降。但值得注意的是,当电凝笔开窗后辅以锁边处理,这一负面效应被有效缓解,拉伸强度未出现显著下降,保持了较好的结构稳定性。

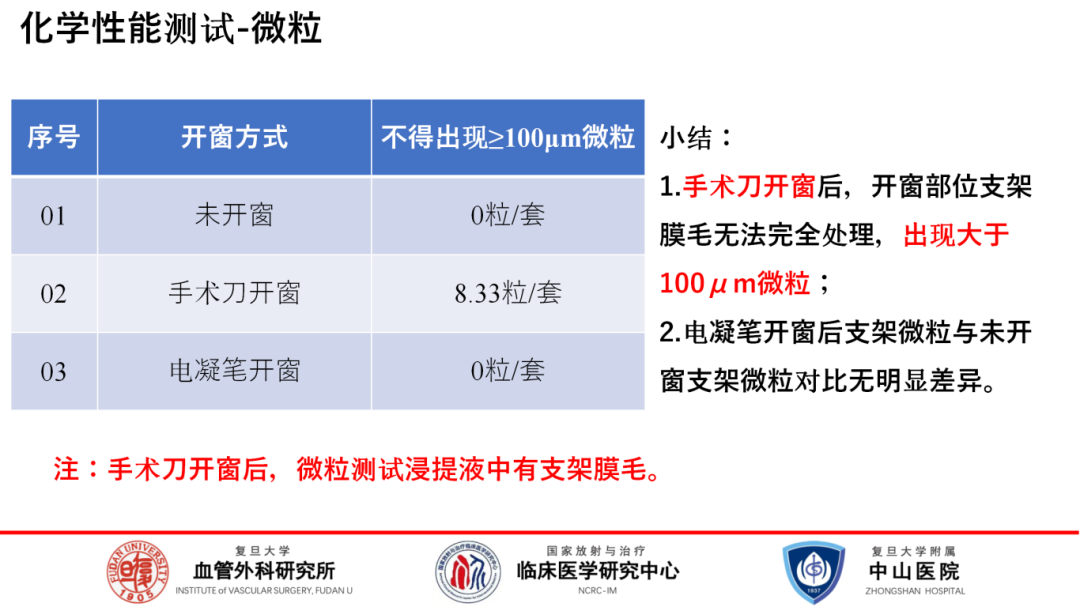
其次,关于开窗操作可能引入的微粒杂质问题。研究发现手术刀开窗后存在大于100μm微粒进入人体循环的风险,而电凝开窗方式则显著降低了这一风险,确保了更高的生物安全性。

综上所述,支架开窗主要影响的是其力学特性,尤其是拉伸强度,而对释放性能及化学性质的影响有限。因此,推荐在临床应用中优先考虑采用电灼开窗并结合锁边技术,这既能最大限度地保留支架的结构完整性和力学强度,又能有效防止微粒污染,确保支架的整体性能及安全性。
国内外相关法规
美国对未获FDA批准的医疗器械,在紧急使用、同情使用或研究性设备豁免(IDE)试验情况下允许使用,但需备案及报告。

相比之下,我国虽无专门针对“改制”器械的法律,但对“自制”器械管控严格,违法生产后果严重。针对“定制支架”,《定制式医疗器械监督管理规定(试行)》明确了企业资质和产品要求。


改制支架不仅涉及结构改变,还涉及超适应症使用。我国《医疗器械临床使用管理办法》等规定医疗机构需遵守器械适用范围,但“超适应证”使用现象普遍,原因包括临床试验局限性、说明书变更难及成本高等。



《中华人民共和国医师法》允许特定条件下超说明书用药,而医疗器械领域尚缺类似专家共识。合规超适应症使用医疗器械可通过注册研究者发起的临床研究(IIT),需满足医院水平、疾病无有效干预、观察性研究提示必要及临床证据安全等条件,与美国IDE有相似之处。

综上,目前我国对PMEG尚未出台针对性法律法规,存在法律风险。因此,为了最大程度降低风险,可从以下三点入手:1、确保充分的患者知情同意;2、严格遵循伦理审查与批件获取;3、将治疗纳入IIT(研究者发起的临床试验)管理。
匹兹堡大学医学中心(UPMC)——覆盖美国五个州,提供了一个集中的转诊系统,具备处理复杂主动脉病变的专业知识和资源。UPMC通过以下步骤构建了复杂主动脉疾病的腔内治疗体系并强化质控:
1、标准化评估与计划:病人转诊至PMEG项目后,经历包括围术期全面评估、个性化病例计划、多学科讨论及超适应症手术知情同意的标准流程。外科医生在所有检查完成前不作出手术决定。
2、处理单点故障:术前进行潜在故障点排查,强调多学科(如心肺评估、抗凝规划等)共同参与,减少单一外科医生决策的风险,特别是测量数据转移到支架过程。UPMC使用3D打印的圆柱模板来减少此类错误,模板数据由外科医生确认两次,在无尘环境中使用可消毒材料由3D打印部门打印,在中央处理部门进行消毒,后才可用于PMEG。
3、围术期多学科协作护理:与麻醉、心内、呼吸科等紧密合作,实施标准化护理,常规应用脑脊液引流及神经电生理监测以降低截瘫风险。
4、质量注册与跟踪:所有PMEG患者纳入纵向质量注册系统,追踪器械构建、植入、围术期情况及长期随访结果,以评估并改进治疗质量。
5、监管合规:鉴于FDA对超适应症器械使用的鼓励态度,UPMC已提交IDE申请并积极跟进审批流程,确保合规操作。
总结
综上所述,未来较长时期内,PMEG无疑将持续作为国内治疗复杂主动脉病变的主导策略。借鉴国际先进经验,并紧密结合我国法律法规框架,我们应在IIT的严谨框架下,不断深化PMEG技术流程的规范化建设,实现质控手段与标准的全国统一,为PMEG技术的有效性与安全性构筑坚实的临床证据基础,进而推动形成一套具有中国特色的、高效且安全的复杂主动脉病变腔内治疗“中国方案”。长远来看,这一方案的成熟与成功实施,还将有力促进国家层面相关法律法规的健全与完善,为医疗技术的持续创新与患者福祉的不断提升贡献力量。
